KCNQ4 Anticorps
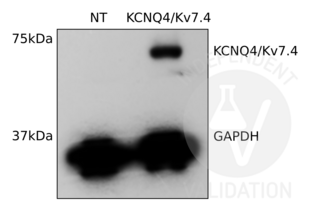
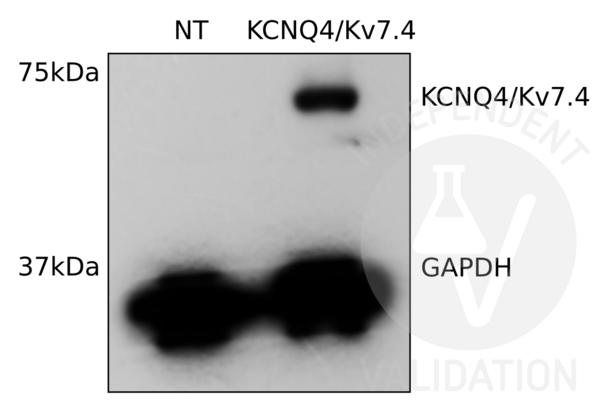 KCNQ4 anticorps (C-Term) (ABIN3185286)
KCNQ4 anticorps (C-Term) (ABIN3185286)
KCNQ4 Reactivité: Humain, Souris WB, ELISA Hôte: Lapin Polyclonal unconjugated
KCNQ4 Reactivité: Humain WB, IHC, IP, IF, ICC, AA Hôte: Souris Monoclonal N43-6 (Formerly S43-6) unconjugated
KCNQ4 Reactivité: Humain WB, ELISA Hôte: Souris Monoclonal 2H6 unconjugated
KCNQ4 Anticorps par Grade
On trouve ici des KCNQ4 Anticorps avec un Grade spécifique. Les Grade mentionnés ici sont quelques-uns de ceux qui sont disponibles. Un clic sur le lien correspondant permet d'accéder aux produits
KCNQ4 Anticorps par Réactivité
Trouvez KCNQ4 Anticorps pour une variété d'espèces telles que anti-Human KCNQ4, anti-Mouse KCNQ4, anti-Rat KCNQ4. Les espèces listées ci-dessous sont parmi celles disponibles. Cliquez sur un lien pour accéder aux produits correspondants.
KCNQ4 Anticorps par Hote
On trouve ici des KCNQ4 Anticorps avec un Hote spécifique. Les Hote mentionnés ici sont quelques-uns de ceux qui sont disponibles. Un clic sur le lien correspondant permet d'accéder aux produits
KCNQ4 Anticorps par Clonalité
Trouvez les KCNQ4 Anticorps monoclonaux ou polyclonaux disponibles. Cliquez sur un lien pour accéder aux produits correspondants.
KCNQ4 Anticorps par Clone
On trouve ici des KCNQ4 Anticorps avec un Clone spécifique. Les Clone mentionnés ici sont quelques-uns de ceux qui sont disponibles. Un clic sur le lien correspondant permet d'accéder aux produits
KCNQ4 Anticorps fréquemment utilisés
- (1)
- (1)
- (3)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (1)
- (1)
- (3)
Dernières publications pour nos KCNQ4 Anticorps
: "Guanylyl Cyclase A/cGMP Signaling Slows Hidden, Age- and Acoustic Trauma-Induced Hearing Loss." dans: Frontiers in aging neuroscience, Vol. 12, pp. 83, (2020) (PubMed).: "Hair cell maturation is differentially regulated along the tonotopic axis of the mammalian cochlea." dans: The Journal of physiology, Vol. 598, Issue 1, pp. 151-170, (2020) (PubMed).
: "GC-B Deficient Mice With Axon Bifurcation Loss Exhibit Compromised Auditory Processing." dans: Frontiers in neural circuits, Vol. 12, pp. 65, (2019) (PubMed).
: "Expression and function of Kv7.4 channels in rat cardiac mitochondria: possible targets for cardioprotection." dans: Cardiovascular research, Vol. 110, Issue 1, pp. 40-50, (2016) (PubMed).
: "Role of Kv7 channels in responses of the pulmonary circulation to hypoxia." dans: American journal of physiology. Lung cellular and molecular physiology, Vol. 308, Issue 1, pp. L48-57, (2015) (PubMed).
: "K(V)7.4 channels participate in the control of rodent renal vascular resting tone." dans: Acta physiologica (Oxford, England), Vol. 214, Issue 3, pp. 402-14, (2015) (PubMed).
: "Kv 7 positive modulators reduce detrusor overactivity and increase bladder capacity in rats." dans: Basic & clinical pharmacology & toxicology, Vol. 110, Issue 2, pp. 145-53, (2014) (PubMed).
: "KCNQ and KCNE potassium channel subunit expression in bovine retinal pigment epithelium." dans: Experimental eye research, Vol. 116, pp. 424-32, (2014) (PubMed).
: "The endocannabinoid 2-AG controls skeletal muscle cell differentiation via CB1 receptor-dependent inhibition of Kv7 channels." dans: Proceedings of the National Academy of Sciences of the United States of America, Vol. 111, Issue 24, pp. E2472-81, (2014) (PubMed).
: "Specification of skeletal muscle differentiation by repressor element-1 silencing transcription factor (REST)-regulated Kv7.4 potassium channels." dans: Molecular biology of the cell, Vol. 24, Issue 3, pp. 274-84, (2013) (PubMed).
Pseudonymes pour KCNQ4 Anticorps
potassium voltage-gated channel subfamily Q member 4 (KCNQ4) Anticorpspotassium channel, voltage gated KQT-like subfamily Q, member 4 (kcnq4) Anticorps
potassium voltage-gated channel subfamily Q member 4 (Kcnq4) Anticorps
potassium voltage-gated channel, subfamily Q, member 4 (Kcnq4) Anticorps
DFNA2 Anticorps
DFNA2A Anticorps
k(v)7.4 Anticorps
KCNQ4 Anticorps
kv7.4 Anticorps
KV7.4 Anticorps
Avez-vous cherché autre chose?
- KCNQ3 Anticorps
- KCNQ2 Anticorps
- KCNQ1 Anticorps
- KCNN4 Anticorps
- KCNN3 Anticorps
- KCNN2 Anticorps
- KCNN1 Anticorps
- KCNMB4 Anticorps
- KCNMB3 Anticorps
- KCNMB2 Anticorps
- KCNMB1 Anticorps
- KCNMA1 Anticorps
- KCNK9 Anticorps
- KCNK7 Anticorps
- KCNK6 Anticorps
- KCNK4 Anticorps
- KCNK3 Anticorps
- KCNK2 Anticorps
- KCNK18 Anticorps
- KCNK17 Anticorps
- KCNQ5 Anticorps
- KCNRG Anticorps
- KCNS1 Anticorps
- KCNS2 Anticorps
- KCNS3 Anticorps
- KCNT1 Anticorps
- KCNT2 Anticorps
- KCNU1 Anticorps
- KCNV1 Anticorps
- KCNV2 Anticorps
- KCP Anticorps
- KCT2 Anticorps
- KCTD1 Anticorps
- KCTD10 Anticorps
- KCTD11 Anticorps
- KCTD12 Anticorps
- KCTD13 Anticorps
- KCTD15 Anticorps
- KCTD18 Anticorps
- KCTD4 Anticorps



